Principal Scientific Researcher
Considered the founder of the industry, Genentech, now a member of the Roche Group, has been delivering on the promise of biotechnology for more than 40 years. Genentech is a biotechnology company dedicated to pursuing groundbreaking science to discover and develop medicines for people with serious and life-threatening diseases. Our transformational discoveries include the first targeted antibody for cancer and the first medicine for primary progressive multiple sclerosis.
Peak Picking, Data QC and TargetedMSQC
Identifying quality peaks in any omics dataset is important not just identify existince of
protein/peptides but also separate noise and signal. To be able to classify peak which not only
of good intensity but also follow the rules of similarity, shape and other features as defined.
MSnbase and XCMS two packges help deal with this problem in a rule based method which can be
followed to an extent but manual intervention is required for more intensive tasks especially
DIA data acquisition methods are used. Increasing sample sizes also demand the use of tools that
require minimal to no manual intervention and a quick turnaround time in terms human interpretable results
through either visuals or summary tables.
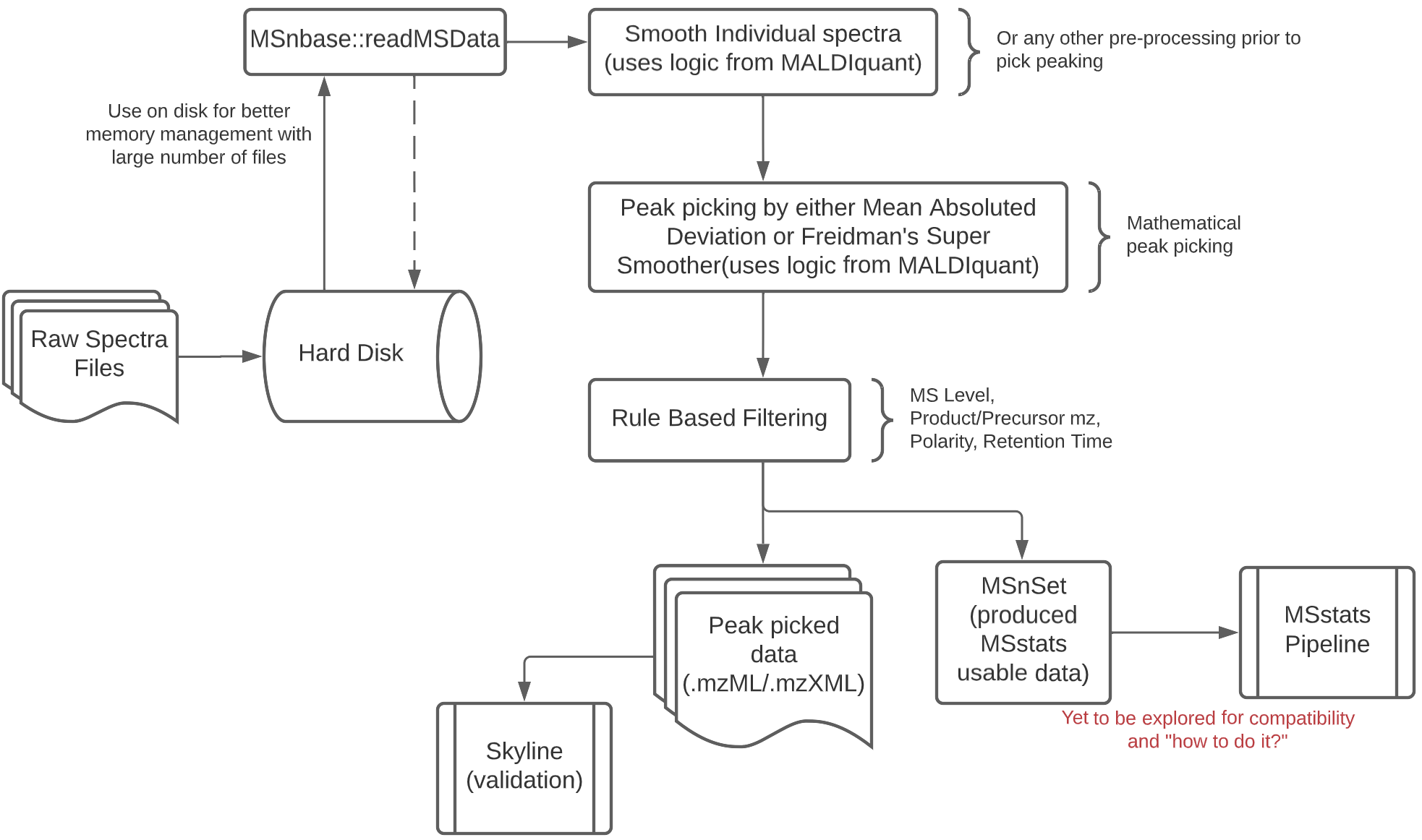
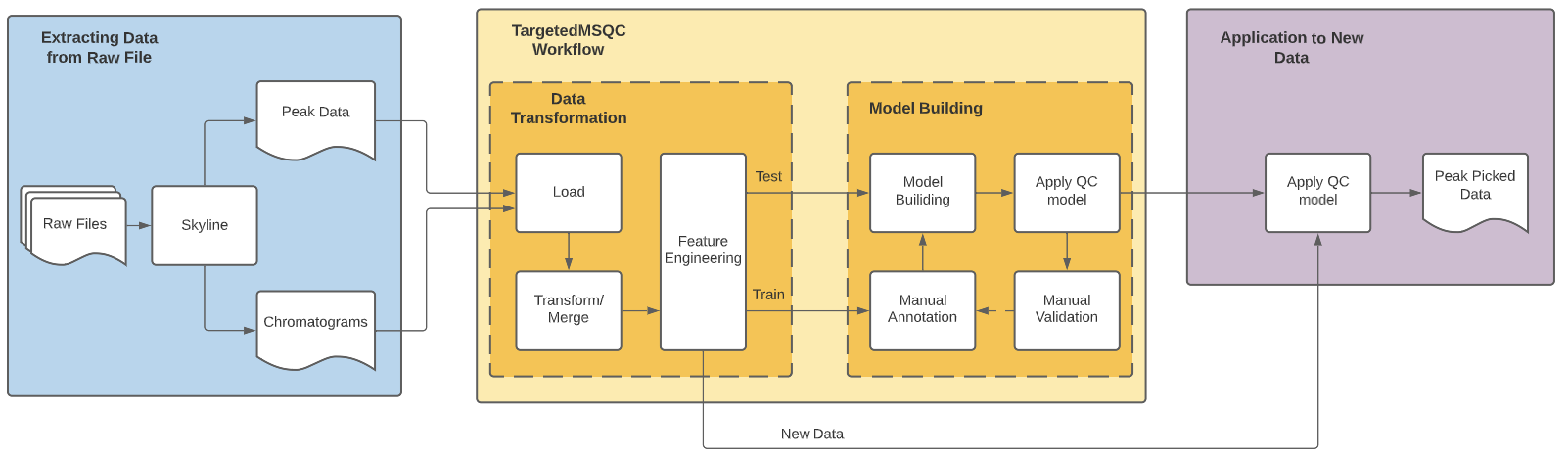
TargetedMSQC was a package developed in-house at Genentech and published for through the paper Quality assessment and interference detection in targeted mass spectrometry data using machine learning the package enables us to use supervised machine learning methods to classify peak quality. My work involved re-factoring the package, do feasibility study around extending a use case for DIA methods and develop an interface to interact with the results without needing to code.
Large sample size and MSstats
MSstats suite of statistical packages help lab based and data scientists analyse Mass Spec proteomics data generated by various tools like Spectronaut, Skyline, DiaNN to name a few. These tools
work very efficiently on limited number of samples very well. One such analytical project involved analysing a CSF dataset with close to 150 samples with data collected using the DIA method resulting
in close to 8000 proteins with multiple fragment ion, differing charge states and having a heavy (reference) and light (target) state. The resulting data file generated by intermediate tools very over
~20GBs and a newer CSF study with 300 samples to ~105GBs.
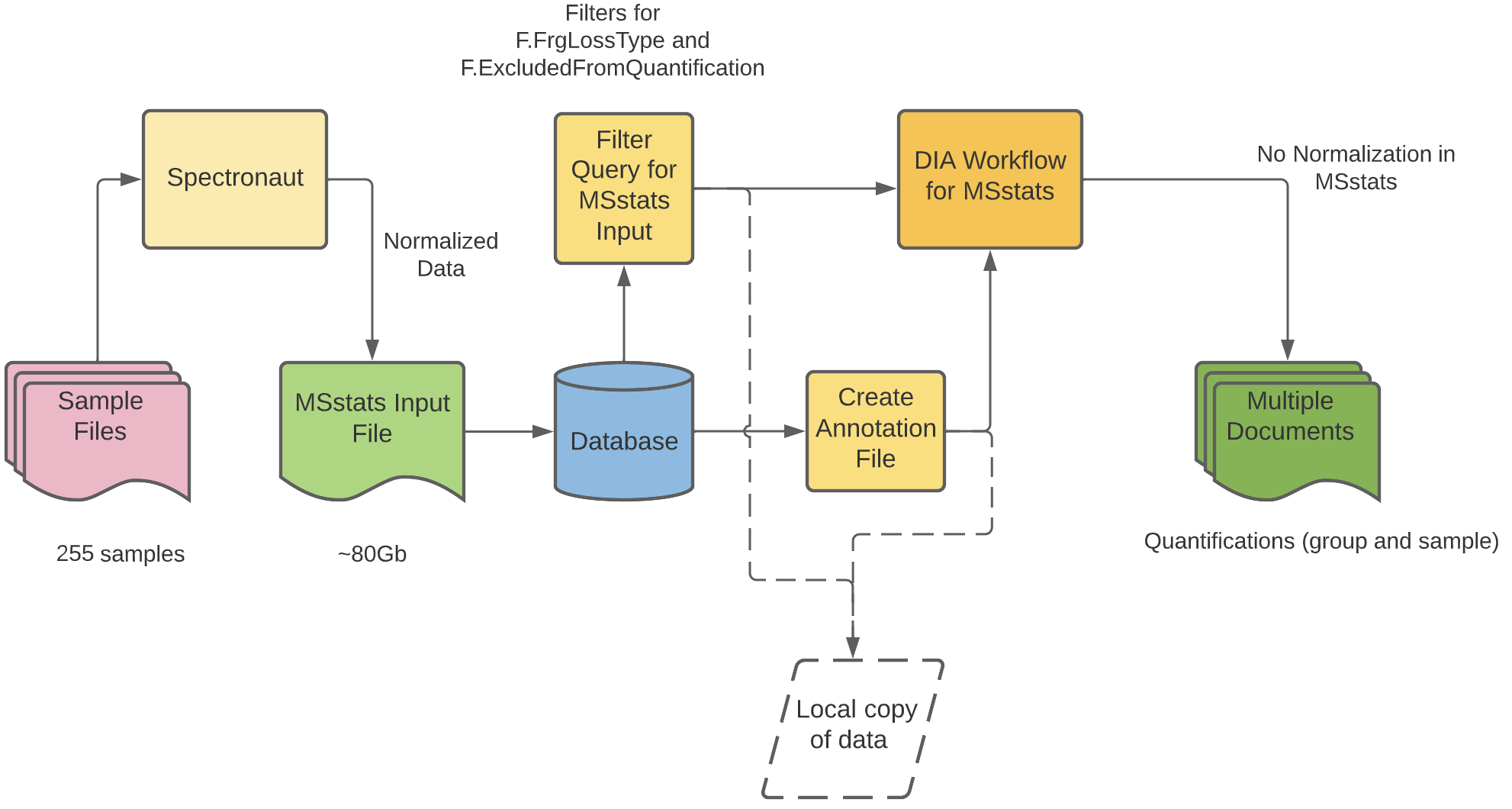
The Image above shows a preliminary workflow to get around the memory limitations of using in memory computations. Stiching together a host of analytical and data manipulation pipelines to generate an analysis ready dataset. This initial workflow also resulted in a further collaboration with the Vitek Lab at Northeastern Univeristy, Boston. A new package called MSstatsBig was developed to allow manipulation of out of memory datasets
Publications, Collaborations & Posters
- Using SILAC to Develop Quantitative Data-Independent Acquisition (DIA) Proteomic Methods
- MSstats Version 4.0: Statistical Analyses of Quantitative Mass Spectrometry-Based Proteomic Experiments with Chromatography-Based Quantification at Scale
- Using stable isotope labeling by amino acids in cell culture (SILAC) to improve data independent acquisition (DIA) relative quantification
- Identifying biomarkers of fibrosis from the urinary proteome of renal fibrosis patients using DIA mass spectrometry
Recommendations